

Calcium is one of the most talked about ions in reef tanks. It is critical for the formation of a variety of structures, including the skeletons and shells of many corals and other organisms. This article is the first of several that will provide a detailed chemical understanding of calcium in seawater, calcium in supplements of various kinds, calcium in biological structures, calcium in various types of rock and sand, the effect of calcium on other ions like phosphate, how organisms take up and deposit calcium, and the monitoring of calcium in tanks. This article will focus on the nature of calcium in seawater and in our tanks, and will answers such questions as “Why does that solid coat my heater?” and “Why does my sand dissolve?”.
Calcium in Seawater
Calcium is one of the major ions in seawater. It is present in seawater at about 410 ppm the world over, so it comprises just under 1.2% by weight of the solids. Variations from this concentration are most often caused by changes in salinity, where the calcium goes up and down just as the salinity does. Another cause for variation comes about from the input of rivers, which are often greatly enriched in calcium relative to other ions such as sodium. Calcium is also frequently enriched in hydrothermal vent water that is released on the bottom of the ocean. The calcium is dissolved from the hot basalt as the water passes through it, and is released to the ocean.
One way to think about the calcium in seawater is to consider how long a typical calcium ion is free in solution before precipitating as a solid such as calcium carbonate. By comparing the known seawater concentration and the amount added by rivers and vents, one can estimate the typical residence time for calcium in the ocean. Likewise, from the seawater concentration and the known sedimentation rates in the ocean, one can do the same thing. Together, these methods produce an estimate of a few million years for the residence time of calcium. By comparison, an ion with little turnover like sodium has a residence time of hundreds of millions of years, and an ion that is quickly removed from the water column, such as aluminum, has a residence of about a thousand years.
Of course, the residence time for calcium in reef tanks is much lower (as low as a week or less for some tanks), and that’s why we are so interested in it: if we don’t keep adding it the concentration soon drops. The same cannot be said for the ocean. If all input of calcium to the ocean stopped today, we’d wouldn’t notice the drop in calcium for a long, long time. The reason for this difference simply boils down to the very small volume of water in our tanks coupled with a high rate of calcium carbonate deposition compared to the huge volume of water in the ocean coupled with a not so high rate of deposition overall.
One interesting aspect of calcium in seawater is that the calcium concentration can be higher in deep ocean water than in surface water. In the Pacific, for example, deep ocean water contains about 1% more calcium than surface water. The reason for this increase is that calcium carbonate becomes much more soluble at high pressure, preventing precipitation of calcium carbonate, and even permitting dissolution of calcium carbonate particulates formed higher in the ocean that settle into the depths.
The reason for this solubility change with pressure is fairly esoteric. When calcium and carbonate ions are dissolved in water, a number of water molecules become tightly attached to the ions. This hydration is discussed in detail in the following section. Overall, however, the volume occupied by undissolved calcium carbonate and water is larger than the volume occupied by calcium and carbonate ions dissolved in this same amount of water. This volume change is primarily due to the increased “density” that can be attained by water molecules around ions compared to pure water. When this dissolution is taking place under the tremendous pressures on the bottom of the ocean, the dissolution can actually be driven by the pressure, because of the volume change involved, resulting in greater solubility.
Finally, calcium in the ocean can be locally depleted in places where precipitation of calcium carbonate is especially rapid. This includes the Bahamas Banks (where oolitic aragonite is precipitated), in parts of the Red Sea, and presumably in some lagoons where calcification is high and the water volume is small.
Chemical State of Calcium in Seawater
In fresh water below pH 11 or so, calcium ions are essentially free. That is, they are not
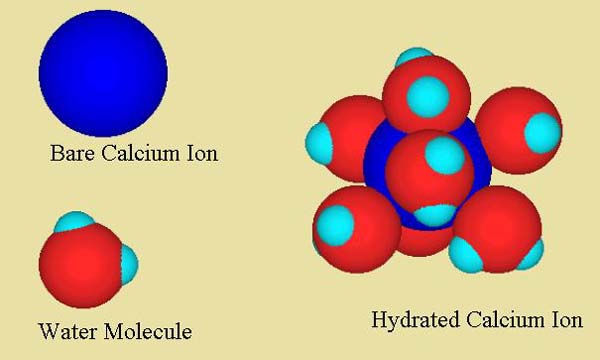
Figure 1. A hydrated calcium ion showing it’s inner sphere of hydrating water molecules. Calcium is shown in dark blue, oxygen in red, and hydrogen in light blue.
strongly attached to anything else except water and move about independently of all other ions in the solution (exceptions being if the water has certain calcium-complexing agents in it, such as phosphate or certain organics). Figure 1 shows a calcium ion hydrated with water molecules. This hydration sphere is quite strongly attached to the ion in water, with about 6-7 water molecules tightly attached. Beyond this first hydration sphere, of course, are other water molecules in a looser arrangement, and beyond that, all of the other things in solution. All of these water molecules around the ion are very rapidly exchanging, but those closest to the ion exchange more slowly and move with it as it moves through the solution.
In seawater, the situation is slightly more complicated. While the majority of calcium ions are still free, some (about 10-15%) are present as an ion pair with sulfate, forming the neutral ion pair CaSO4 (Figure 2). These types of soluble ion pairs are short lived, forming and breaking apart quite rapidly. Nevertheless, they can have significant impact on the properties of seawater. This ion pair is in turn hydrated with water molecules, as shown in Figure 2.
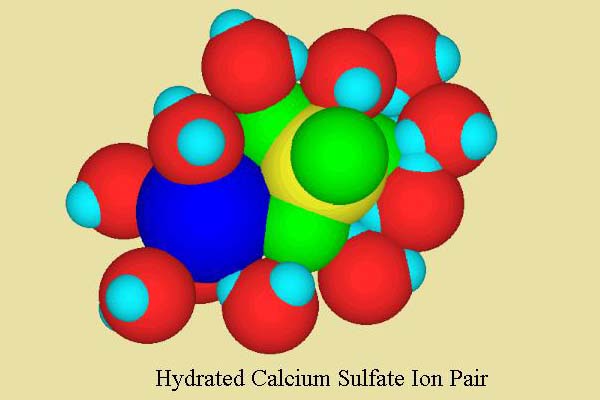
Figure 2. A calcium/sulfate ion pair showing its inner sphere of hydrating water molecules. The colors are the same as Figure 1 except that the sulfur is shown in yellow, and the sulfate oxygen atoms are shown in green to make them stand out from the water molecules. Water molecules have been removed from the top to permit better visualization of the calcium and the sulfate.
Calcium similarly forms ion pairs with carbonate and bicarbonate. While these comprise a small fraction of the total calcium, the calcium carbonate ion pair comprises a fairly large portion of the total carbonate (together with magnesium, about 2/3 of the carbonate). These ion pairs consequently tend to lower the free concentration of carbonate, and thereby help to inhibit precipitation of calcium carbonate, and consequently increase its
solubility.
Finally, calcium forms ion pairs with fluoride, hydroxide, borate, the various forms of phosphate, and other ions to smaller extents that are unimportant to the free calcium concentration, but may impact the free concentrations of these other ions (especially phosphate, where calcium binds to more than 70% of the PO4—). In almost all cases, however,
the effect of calcium is smaller than the effect of magnesium on these ions, both because the concentration of magnesium is higher, and because in some cases it actually interacts more strongly (MgF+ compared to CaF+, for example).
Chemical State of Calcium in Tank Water
Calcium in reef tank water is expected to be similar to that in seawater, with a couple of exceptions. In particular, calcium is readily complexed by many organics, and if present, may form chelates. Some of these are natural (such as carbohydrates and proteins) and may be present in tanks at levels greatly exceeding that in the ocean. Additional calcium-binding agents are sometimes added by aquarists, either intentionally or not. Such compounds include EDTA, citric acid, vitamin C (ascorbic acid), polygluconate, and polyphosphates. Consequently, some of the calcium in reef tanks is expected to be bound to organic compounds. How much and of what type will vary from tank to tank, and may not even be important in most tanks.
Calcium Carbonate in Seawater
One very important aspect of calcium is that, in seawater, it is actually supersaturated. What supersaturation means in this context is that given the right circumstances, it will precipitate as solid calcium carbonate. Of course, under many other circumstances, it doesn’t do so, and the question of why is very important. First, some definitions.
The equilibrium constant expression for the dissolution of calcium carbonate is shown below:
(1) K = [Ca++][CO3—]
When K = Ksp* (the solubility product constant in seawater at any given temperature, pressure, and salinity), then the solution is said to be exactly saturated (equation 2).
(2) Ksp* = [Ca++][CO3—] (saturation)
When the product of the concentration of calcium and carbonate exceeds the Ksp*, the solution is said to be supersaturated, and there is “too much” calcium and carbonate in solution (equation 3).
(3) Ksp* < [Ca++][CO3—] (supersaturation)
When the product of the concentration of calcium and carbonate is less than the Ksp*, the solution is said to be undersaturated, and calcium carbonate can dissolve if put into the solution (equation 4).
(4) Ksp* > [Ca++][CO3—] (undersaturation)
In normal seawater, equation 3 holds (supersaturation). The product of calcium and carbonate is about 3 times the Ksp* of aragonite and 5 times that of calcite (aragonite and calcite are different crystal forms of calcium carbonate; these differences will be discussed more in future articles). Consequently, calcium carbonate is poised to precipitate from seawater, given the opportunity.
When Can Calcium Carbonate Precipitate from Seawater?
One situation in which calcium carbonate can precipitate involves adding calcium carbonate seed crystals of some type to seawater. In many cases, this action will initiate precipitation of calcium carbonate (and magnesium as well). This precipitation typically does not proceed until all of the supersaturation is gone, but is stopped by some other processes (see below).
A second situation where precipitation takes place is if the supersaturation is pushed to unusually high levels. This can be caused by a rise in pH, a rise in temperature (discussed below), or more obviously, by a rise in either calcium or carbonate.
After some solid calcium carbonate has entered the system (either the ocean or a tank), either by precipitation or by addition by the aquarist (e.g., calcium carbonate sand), precipitation will begin immediately. Interestingly, several things stop the precipitation in seawater, permitting the ocean to be supersaturated. Without these processes, it is unlikely that the ocean could remain supersaturated, and might even make it impossible for corals to maintain skeletons without expending considerable effort to prevent dissolution.
What processes inhibit continued precipitation of CaCO3 onto a growing crystal? The main thing happening in normal seawater is likely the impact of magnesium. It does two critical things (discussed in detail in this linked article):
- Magnesium holds onto carbonate ions and reduces their free concentration, thereby reducing the likelihood of precipitation onto calcium carbonate surfaces.
- Magnesium gets onto the growing surface of the crystal, essentially poisoning it for further precipitation of calcium carbonate.
Note that while both of these processes inhibit precipitation of calcium carbonate, the first actually increases the solubility, whereas the second does not. It is worth noting that the solubility of calcium carbonate in seawater is about 26 times higher than in freshwater at the same temperature, and this first effect of magnesium is one of the reasons. The second process above does not make calcium carbonate any more soluble. In a sense, it inhibits the pathway between soluble calcium and carbonate ions, and the solid calcium carbonate that would
form.
Other processes that inhibit crystal growth in reef tanks involve both phosphate and organics that get onto the growing crystal, inhibiting it just as magnesium does. Here’s a link to some data on the binding of phosphate to aragonite surfaces. These processes presumably happen in natural seawater as well, but since the concentrations of both organics and phosphate may be higher in tanks, their effect may be enhanced. There is an extensive discussion of these issues in “Captive Seawater Fishes” by Stephen Spotte (1992).
Solubility of Calcium Carbonate in Marine Aquaria
There are several interesting results of the supersaturation of calcium carbonate in our tanks. One that many people quickly encounter in maintaining a reef tank is that heaters and other warm objects (e.g., pump impellers) seem to become coated with solids over time. Why is this?
This solid is principally calcium carbonate, though it likely has other ions in the crystal as well (magnesium and other metals, phosphate and other anions, etc.). The two reasons that this happens are readily understood, but the primary one is not at all obvious.
One contribution to the reason that calcium carbonate precipitates on heaters is simply that calcium carbonate in seawater is slightly less soluble as the temperature rises. Since calcium carbonate is already supersaturated, the effect is that when the water is warmed, the supersaturation of calcium carbonate rises, making precipitation more likely.
The supersaturation (W) for calcium carbonate in seawater is given by:
(5) W = [Ca++][CO3—]/Ksp*
When W = 1, the solution is saturated, and when W >1, the solution is supersaturated. The higher W is, the more likely precipitation is to take place. At S=35 and 1 atmosphere pressure, the Ksp* decreases slightly as the temperature rises. Millero (“Chemical Oceanography”, 1996) provides a series of long equations for calculating Ksp* for both aragonite and calcite. For aragonite, the log Ksp* drops from -6.19 at 25 °C, to -6.23 at 40 °C to -6.44 at 80 °C. In relative terms, the Ksp* has gone from 1 to 0.91 to 0.55 over this temperature range. Likewise for calcite, the relative Ksp* has changed from 1 to 0.96 to 0.73 over this range.
Consequently, if a tank has a supersaturation of about 3 for aragonite and 5 for calcite at 25 °C (typical for seawater), then at 40 °C the supersaturation has increased to about 3.3 and 5.2, respectively. At 80 °C this supersaturation has increased to 5.4 and 6.8, respectively. Since the supersaturation has increased, the likelihood of precipitation has increased, and this increase is part of the explanation of why precipitation takes place on heaters.
Bicarbonate Acidity Shift with Temperature
A second, and perhaps unexpected contribution to the precipitation of calcium carbonate on warm objects has to do with the concentration of carbonate. As water is heated, the equilibrium between bicarbonate and carbonate (equation 6) is shifted toward carbonate.
(6) HCO3– <– –> H+ + CO3—
(7) Ka* = [CO3—][H+]/[HCO3–]
(8) [CO3—] = Ka*[HCO3–]/[H+]
(9) pKa* = -log Ka*
This shift toward carbonate is evidenced by the shift in the seawater pKa* for bicarbonate from 9.00 at 25 °C to 8.68 at 40 °C and 8.16 at 80 °C (calculated from equations provided by Millero; the * simply indicates that it is in seawater at a given temperature, pressure, and salinity).
From equation 8 (and related derivations), we see that if the Ka* rises, then [CO3—] will rise, [H+] will rise, and [HCO3–] will decline. The question is by how much.
Assuming that the carbonate concentration is much less than the bicarbonate concentration, we can determine the change in H+ with equation 10. Equation 10 is simply the solution of equation 8 for H+:
(10) [H+] ~ [Ka*C + Kw*]1/2
where C is the total concentration of carbonate/bicarbonate/carbonic acid species and pKw* is the constant for the autodissociation of water. [About the assumption that the carbonate concentration is less than the bicarbonate concentration: we know this is true for seawater at 25 °C, but it is also generically shown by Pankow (“Aquatic Chemistry Concepts”, 1991; p. 84) to be valid with this combination of pKa* (about 9), pKw* (about 13) and C (about 2 mM) for other temperatures as well].
Using the values of Ka* and Kw* at the appropriate temperatures, we find that [H+] has increased by a factor of about 1.45 between 25 and 40 °C. As a point of curiosity, this has resulted in a decrease in pH of about 0.16 units.
Still, what we want to know is the change in the carbonate concentration. Going back to equation 8 we have:
(11) [CO3—]25 = Ka*25 [HCO3–]25/[H+]25
for 25 °C and
(12) [CO3—]40 = Ka*40 [HCO3–]40/[H+]40
for 40 °C. We assume, as above, that
[HCO3–]25 = [HCO3–]40 (that is, that the bicarbonate concentration is so high that taking a bit away to form carbonate does not impact the bicarbonate concentration significantly). Substituting the known change in Ka* (Ka*40 = 2.1Ka*25 ) and H+ ([H+]40 = 1.45[H+]25 ), we get
(13) [CO3—]40 = (2.1Ka*25)[HCO3–]40/1.45[H+]25
Combining equations 11 and 13, and the fact that
[HCO3–]40 ~ [HCO3–]25 , we get
(14) [CO3–]40 = 1.45 [CO3—]25
Consequently, on going from 25 °C to 40 °C, the relative concentration of carbonate has increased by a factor of 1.45. We can now go back and confirm our assumption that carbonate is still far below the bicarbonate concentration, and it clearly is, so that assumption was valid.
Going back to what we really care about, the supersaturation of calcium carbonate, we find that if the carbonate has increased by a factor of 1.45, then the supersaturation of both calcite and aragonite have increased by the same factor (equation 5).
Running the same calculations for 80 °C (pKa = 8.16) we get the carbonate concentration to increase by a factor of 2.4x compared to 25 °C. The [H+] also increases by the same factor.
Comparing Solubility and Acidity Changes on Supersaturation
of Calcium Carbonate
Comparing the increase in supersaturation due to solubility and acidity changes between 25 and 40 °C for aragonite, we find a change from W = 3.0 to 3.3 due to solubility, and from 3.0 to 4.4 due to the bicarbonate pKa shift. Together these effects yield a supersaturation of 4.8 for aragonite.
Similarly for calcite, the solubility change between 25 and 40 °C causes an increases in W from 5.0 to 5.2 for the solubility change and from 5.0 to 7.3 for the bicarbonate pKa shift. Together these effects yield a supersaturation of 7.5 for calcite.
Comparing the increase in supersaturation due to solubility changes between 25 and 80 °C, we find a change from W = 3.0 to 5.4 due to solubility, and from 3.0 to 7.2 due to the bicarbonate pKa shift. Together these effects yield a supersaturation of 13 for aragonite.
Similarly for calcite, the solubility change between 25 and 80 °C causes an increases in W from 5.0 to 6.8 for the solubility change and from 5.0 to 12 for the bicarbonate pKa shift. Together these effects yield a supersaturation of 16.3 for calcite.
What do these values really mean? The following combinations of calcium and alkalinity have the same supersaturation in seawater:
- Normal seawater at 80 °C
- Seawater at 25 °C with calcium raised to 1300 ppm
- Seawater at 25 °C with the alkalinity raised to 8.2 meq/L
It makes sense that all three of the situations above could lead to precipitation of calcium carbonate, and that is exactly what happens on the surfaces of hot objects in our tanks.
Dissolution of CaCO3 in Aquaria
If calcium carbonate is supersaturated in marine aquaria, how can it also dissolve? The answer lies in the fact that while the water column is supersaturated, other parts of the tank may not be. Specifically, the interstitial water of sand and rock if often lower in pH than the water column. For example, if I put a pH probe into my oolitic aragonite sand bed, I get a pH in the 7’s, when the water column itself has pH = 8.4.
The reasons for the pH being lower in the sand are beyond the scope of this article, but it relates to the breakdown of organics (and some nitrogen compounds). Both aerobic and anaerobic oxidation of organics in seawater can lead to the production of acid, especially carbonic acid derived from CO2. This link is to a thread that shows some of the reactions that can (and cannot) produce acid in sand beds.
As the pH is lowered, the equilibrium between carbonate and bicarbonate shifts towards bicarbonate (i.e., shifts to the left in equation 15):
(15) HCO3– <– –> H+ + CO3—
This shift considerably lowers the carbonate concentration. Using equation 8, one can calculate that the concentration of carbonate drops by about a factor of 3 for a pH drop of 0.5 and by a factor of 10 for a full pH unit drop. Consequently, aragonite first becomes soluble in seawater when the pH drops below about 7.7 (this value might be more like 7.5-7.7 in reef tanks where the alkalinity is often higher than in seawater). That level is attained in some sand beds, and permits the dissolution of some of the sand.
The rate of dissolution is fairly low, however, because the rate of delivery and degradation of organics (or certain nitrogen compounds) deep enough in the sand to permit a pH drop is fairly low. The rate will, however, vary from tank to tank as the different ways of delivering organics to deeper parts of the sand will vary (diffusion; movement by organisms; death of organisms, etc). Note that the need to oxidize the organics in deeper parts of the sand to permit dissolution of the sand has nothing to do with the oxygenation of the sand. It has more to do with the fact that at near surface regions of the sand, the pH will be closer to that of the tank water by acid and base transfer from the water column, and you need to be deep enough to permit a lower pH to become established.
Conclusion
So begins the journey to a full understanding of the many aspects of calcium in reef tanks. It is a complicated ion with a variety of critical applications in reef tanks. Having a greater appreciation for some of these applications will not only leave you better informed about how your tank works, but in how to deal with, or even better, prevent problems involving calcium.
Literature References for Further Reading.
- Crystal growth of calcium carbonate in artificial seawater: The effect of temperature and of the presence of inhibitors. Kladi, Elina; Ostvold, Terje; Klepetsanis, Pavlos G.; Amjad, Zahid; Koutsoukos, Petros G. FORTH-ICEHT, Patras, Greece. Book of Abstracts, 218th ACS National Meeting, New Orleans, Aug. 22-26 (1999).
- Solubility of calcite in seawater solution of different magnesium concentrations at 25°C and 1 atm total pressure: a laboratory re examination. Rushdi, Ahmed I.; Chen, Chen-Tung Arthur; Suess, Erwin. Coll. Oceanic Atomospheric Sci., Oregon State Univ., Corvallis, OR, USA. Umi (1998), 36(1), 9-22.
- The problem of alkaline scale formation from a study on Arabian Gulf [Persian Gulf] water. El Din, A. M. Shams; Mohammed, Rizk A. Mater. Test. Lab., Water Electr. Dep., Abu Dhabi, United Arab Emirates. Desalination (1989), 71(3), 313-24.
- Apparent calcite supersaturation at the ocean surface: why the present solubility product of pure calcite in seawater does not predict the correct solubility of the salt in nature. Cooke, Robert C.; Kepkay, Paul E. Dep. Oceanogr., Dalhousie Univ., Halifax, NS, Can. Mar. Chem. (1984), 15(1), 59-69.
- Calculation of saline waters supersaturation on calcium carbonate, magnesium hydroxide and calcium sulfate, taking into account ion associates formation. Martynova, O. I.; Vasina, L. G.; Krotova, I. S. Inst. Power Eng., Moscow, USSR. Proc. Int. Symp. Fresh Water Sea (1976), 1 329-36.
- Calcium carbonate retention in supersaturated sea water. Pytkowicz, R. M. Sch. Oceanogr., Oregon State Univ., Corvallis, Oreg., USA. Amer. J. Sci. (1973), 273(6), 515-22.
- Inhibition of aragonite precipitation from supersaturated sea water: a laboratory and field study. Berner, Robert A.; Westrich, Joseph T.; Graber, Ron; Smith, Jennifer; Martens, Christopher S. Dep. Geol. Geophys., Yale Univ., New Haven, Conn., USA. Am. J. Sci. (1978), 278(6), 816-37.
- Biologically mediated dissolution of calcium carbonate above the chemical lysocline? Milliman, J. D.; Troy, P. J.; Balch, W. M.; Adams, A. K.; Li, Y.-H.; Mackenzie, F. T. School of Marine Science of the Virginia Institute of Marine Science, College of William and Mary, Gloucester Pt., VA, USA. Deep-Sea Res., Part I (1999), 46(10), 1653-1669.
- Excess dissolved Ca in the deep ocean: a hydrothermal hypothesis. De Villiers, Stephanie. Department of Geological Sciences, University of Washington, Seattle, WA, USA. Earth Planet. Sci. Lett. (1998), 164(3-4), 627-641.
- Evidence in support of first-order dissolution kinetics of calcite in seawater. Hales, Burke; Emerson, Steve. Lamont-Doherty Earth Observatory of Columbia University, Palisades, NY, USA. Earth Planet. Sci. Lett. (1997), 148(1-2), 317-327.
- The crystallization of calcium carbonate in artificial seawater; role of the substrate. Sabbides, Theophylaktos G.; Koutsoukos, Petros G. Institute of Chemical Engineering and High Temperature Chemical Processes, and Department of Chemical Engineering, University of Patras, P.O. Box 1414, Patras, Greece. J. Cryst. Growth (1993), 133(1-2), 13-22.
- Possible role of carbonate dissolution in estuarine phosphate dynamics. De Jonge, V. N.; Villerius, L. A. Tidal Waters Div., Rijkswaterstaat, Haren, Neth. Limnol. Oceanogr. (1989), 34(2), 332-40.
- Synthetic magnesian calcites solubilities in seawater as a function of pressure. Koch, B. Lab. Oceanol., Univ. Liege, Sart Tilman, Belg. Oceanol. Acta (1985), 8(4), 413-21.
- Kinetics of dissolution and precipitation of calcium carbonate in seawater. Berner, Robert A. Dep. Geol. Geophys., Yale Univ., New Haven, CT, USA. Crist., Deform., Dissolution Carbonates, Reun. (1980), 33-57.
- The solubility of calcium carbonate in sea water at 2° C based upon in-situ sampled pore water composition. Sayles, Frederick L. Woods Hole Oceanogr. Inst., Woods Hole, MA, USA. Mar. Chem. (1980), 9(4), 223-35.
- The thermodynamics of the carbonate system in seawater. Millero, Frank J. Rosenstiel Sch. Mar. Atmos. Sci., Univ. Miami, Miami, FL, USA. Geochim. Cosmochim. Acta (1979), 43(10), 1651-61.
- The effect of pressure on the solubility of calcite in seawater at 25°C. Millero, Frank J. Rosenstiel Sch. Mar. Atmos. Sci., Univ. Miami, Miami, Fla., USA. Geochim. Cosmochim. Acta (1976), 40(8), 983-5.
- Dissolution kinetics of calcium carbonate in seawater: VII. The dissolution kinetics of synthetic aragonite and pteropod tests. Morse, John W.; De Kanel, John; Harris, Karen. Rosenstiel Sch. Mar. Atmos. Sci., Univ. Miami, Miami, FL, USA. Am. J. Sci. (1979), 279(5), 488-502.
- Dissolution kinetics of calcium carbonate in sea water. VI. The near-equilibrium dissolution kinetics of calcium carbonate-rich deep sea sediments. Morse, John W. Rosenstiel Sch. Mar. Atmos. Sci., Univ. Miami, Miami, Fla., USA. Am. J. Sci. (1978), 278(3), 344-53.
- Dissolution kinetics of calcium carbonate in sea water. V. Effects of natural inhibitors and the position of the chemical lysocline. Morse, John W. Dep. Oceanogr., Florida State Univ., Tallahassee, Fla., USA. Amer. J. Sci. (1974), 274(6), 638-47.
- Dissolution kinetics of calcium carbonate in sea water. IV. Theory of calcite dissolution. Berner, Robert A.; Morse, John W. Dep. Geol. Geophys., Yale Univ., New Haven, Conn., USA. Amer. J. Sci. (1974), 274(2), 108-34.
- Dissolution kinetics of calcium carbonate in sea water. III. New method for the study of carbonate reaction kinetics. Morse, John W. Dep. Oceanogr., Florida State Univ., Tallahassee, Fla., USA. Amer. J. Sci. (1974), 274(2), 97-107.
- Dissolution kinetics of calcium carbonate in sea water. II. Kinetic origin for the lysocline. Morse, John W.; Berner, Robert A. Dep. Geol. Geophys., Yale Univ., New Haven, Conn., USA. Amer. J. Sci. (1972), 272(9), 840-51.
- Dissolution kinetics of calcium carbonate in sea water. I. Saturation state parameters for kinetic calculations. Berner, Robert A.; Wilde, Pat. Dep. Geol. Geophys., Yale Univ., New Haven, Conn., USA. Amer. J. Sci. (1972), 272(9), 826-39.

Thanks for this great overview of calcium chemistry in seawater. Super-helpful and much appreciated.
Hello Randy, this is an excellent summary and could you advise if you have published a paper on seawater calcium carbonate chemistry at any technical conferences and if so any reference to the article ?